Képzeld el, hogy egészségesnek tűnő gyereked nyolc éves kora után előbb tolószékbe kényszerül, mert lábai felmondják a szolgálatot, majd pár évvel később a kezeivel történik ugyanez, magatehetetlenné és kiszolgáltatottá téve őt, s végül huszas éveiben egyszer csak a szíve és/vagy a tüdeje is feladja. És most képzeld el, hogy bár előre tudod mindezt, úgy kell végignézd, hogy semmit nem tehetsz ellene.
Ezt az agóniát kell ma is átélniük világszerete szülők ezreinek, akik gyermekei izomsorvadásban szenvednek, mert lassan évtizedes kutatómunka ellenére igazolt gyógymód ma sincs, amivel a halál ezen, egyénre és családra nézve egyaránt kegyetlen formáját el lehetne kerülni.

Az izomsorvadások egyik legismertebb formája a Duchenne féle izomdisztrófia (DMD) a ritka genetikai betegségek egyik leggyakrabbika (itthon 118 beteg gyereket tartanak nyilván), ennek megfelelően viszonylag nagy figyelem övezte már hosszú évek óta és számos - gyakran beteg gyerekek szülei által létrehozott alapítványok pénzelte - kutatás irányul(t) valamifajta terápia kifejlesztésére.
A betegségben mutáns DMD gén a dystrophin fehérjét kódolja, aminek fontos szerepe van abban, hogy az izomrostokat a sejtmembránhoz horgonyozza. Ha működésképtelenné válik, akkor az fokozatosan az izomsejtek elpusztulásához, vagyis izomsorvadáshoz vezet. A legtöbb káros mutáció azt eredményezi, hogy egy túl rövid és kevésbé jól működő fehérje keletkezik, ráadásul abból is kevesebb, mert a sejt igyekszik a belső minőségellenőrző mechanizmusai által hibásnak ítélt fehérjéből minél kevesebbet gyártani, hiába van ez esetben, hogy még a részlegesen működő fehérje is jobb lenne, mint annak a teljes hiánya.
A napokban három, nagyon hasonló megközelítést alkalmazó cikk is megjelent a Science honlapján, amelyben a legújabb genomszerkesztési módszereket alkalmazva, egy potenciális génterápiát dolgoztak ki a DMD ellen - igaz, egyelőre csak egerekben. Az amerikai kutatók (Harvard, Duke és University of Texas) a korábban már errefele is emlegetett CRISPR technológiát alkalmazva egy olyan mesterséges adenovírust hoztak létre, amely képes az izomsejteket megfertőzni, majd azokban a hibás génszakaszt kivágni. Az "exon-skipping"-nek nevezett eljárás ugyan nem állítja vissza teljesen a működőképes dystrophint, de egy olyan, kevésbé káros mutációt hoz létre, ami elegendő, kicsit hibás, de azért még működőképes fehérje termelődését biztosítja.
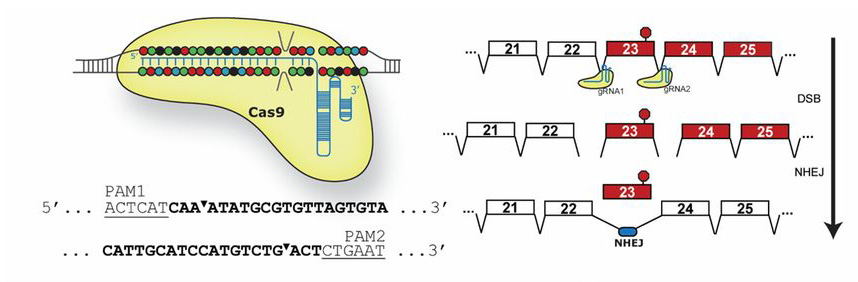
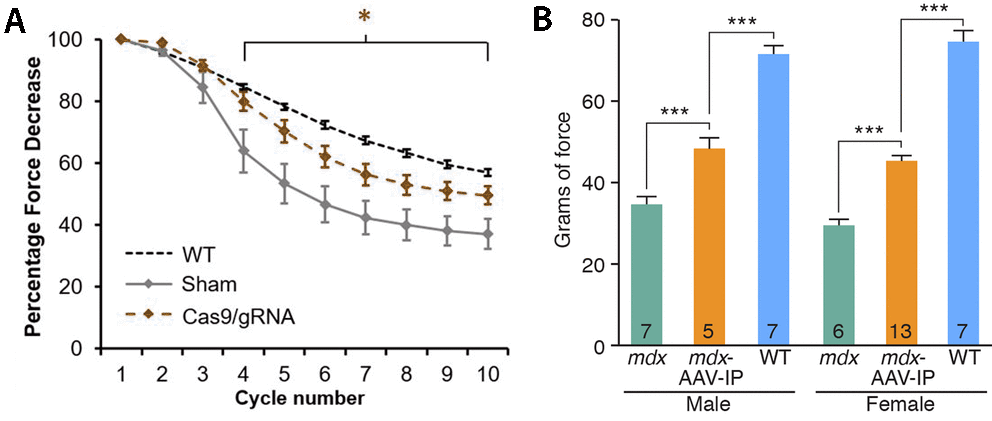
Az izmokba juttatott vírus hatására, pár hónappal a kezelések elkezdése után már jelentősen megugrott a sejtekben jelen levő dystrophin fehérje mennyisége. És bár ugyan még így is csak kb. tizede lesz az egészséges sejtekben levőének, ez már elegendő ahhoz, hogy a kezelt egerek az erőnléti teszteken is sokkal jobban teljesítsenek, mint kezeletlen társaik.
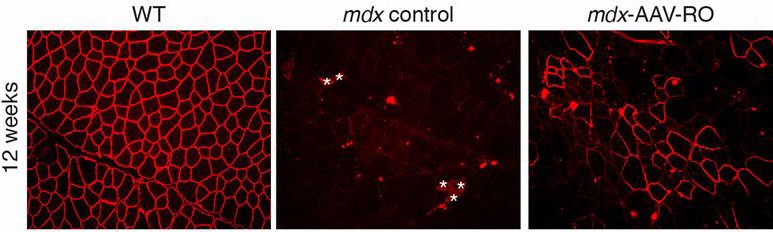
Az exon-skipping alapú eljárás egyébként nagyon hasonló logikán alapul, mint amit két, eddig ígéretesnek tartott DMD gyógyszerjelölt is alkalmazott, ahol szintetikus molekulák segítségével próbálták elérni, hogy a sejtben a DMD gén expressziója során a hibás géndarabot ne vevődjön figyelembe.
A drisapersent and eteplirsent már hosszú évek óta fejlesztik és rengeteg, már-már csodával határos anekdotikus gyógyulási sztori fűződik a nevükhöz, aminek következtében a Duchenne-os gyerekek családjai a Texas Buyers' Club-ban látott elszántsággal és lelkesedéssel lobbiztak, hogy saját gyermekük bekerüljön egy vizsgálati csoportba, s ugyanakkor azért is mindent megtettek, hogy a szövetségi gyógyszerengedélyezésért felelős Food and Drug Administration-nel (FDA) hatásosnak fogadja el ezeket az anyagokat (ami megnyitotta volna az utat a gyógyszerek kereskedelmi forgalmazása előtt). Az újságbeszámolók alapján egészen ígéretesnek tűnő klinikai kísérletek azonban nem bizonyultak elegendőnek, hogy minden kétséget elűzzenek a két szer hatásossága felől (vagyis, hogy placebonál erősebb a hatásuk), így a napokban előbb a drisapersen, majd az eteplirsen engedélykérelmét is visszautasította az FDA.
Mindez még fontosabbá teszi a fent ismertetett CRISPR-alapú megközelítést, aminek egyébként az előnye, hogy a két gyógyszerrel ellentétben, nem igényel folyamatos terápiát, hiszen meghatározott számú kezelés az összes releváns izomsejtnek átírja a genomját, így ezekben csak a dystrophin exon-hiányos, de működő variánsa keletkezik egy idő után.
Kérdés azonban, hogy míg a szükséges klinikai vizsgálatokhoz való előkészületek megtörténnek, illetve a beszerzendő engedélyek megérkeznek, mihez fognak kezdeni azok, akik eddig ebbe a két szerbe tették minden reményüket? És persze az is, hogy egy CRISPR-alapú terápia milyen hamar futhat át az FDA-n.
Óvatosságra ad okot, hogy a korábbi génterápiás fellángolás, a kétezres évek elején, végül egy tragikus próbálkozás miatt nagyon gyorsan kihunyt. Igaz, ez esetben, a korábbi eljárással ellentétben nem a genom egy véleltlenszerű helyére akarnak beilleszteni valamit, hanem egy jól meghatározott helyéről kivágni, ami nagyon-nagyon lecsökkenti annak az esélyét hogy nem várt mellékhatás legyen. Mivel a DMD családoknak nincs igazán veszteni valójük - a betegség kiszámíthatóan végzetes, sajnos, így az idő ellenük dolgozik - biztos lesz elegendő önkéntes alany a klinikai tesztekre, az FDA-n pedig nagy lesz a nyomás, hogy ezt a reménysugarat érdemben megvizsgálja. Remélhetőleg pozitívabb kimenettel, mint a korábbiakat.
(Kapcsolódik még a Statnews és a MosaicScience cikke.)
[A poszt eredetileg a ScienceMeetup blogjában jelent meg.]
Long C, Amoasii L, Mireault AA, McAnally JR, Li H, et al. (2016) Postnatal genome editing partially restores dystrophin expression in a mouse model of muscular dystrophy. Science 351: 400-3.
Nelson CE, Hakim CH, Ousterout DG, Thakore PI, Moreb EA, et al. (2016) In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science351: 403-7.
Tabebordbar M, Zhu K, Cheng JK, Chew WL, Widrick JJ, et al. (2016) In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351: 407-11.